我组新引进李唐博士报导了首个酶与低反应活性的卤代烷衍生物之间的N-取代反应的分子机制,该成果发表在J Phys Chem Lett上。
(https://pubs.acs.org/doi/10.1021/acs.jpclett.8b02225)
在酶抑制剂研发领域,共价抑制剂通常比非共价抑制剂具有更多优势,例如较低的耐药性、抑制效果更持久、较低的给药量以及较少的副作用等。尽管有较多的优势,较高的毒性却是大多数上一代共价抑制剂所无法避免的问题,因为所使用的反应基团通常具有很高的活性和较低的特异性,并且时常导致免疫原性反应。因此设计具有较低的反应活性,但具有高选择性的共价抑制剂具有很重要的研究和应用价值。
多数共价抑制剂都是基于半胱氨酸巯基的反应活性,但由于半胱氨酸不常出现在酶的活性中心,或者其在活性中心的位置不容易被触及,研究者们也尝试了许多别的氨基酸残基,例如赖氨酸、丝氨酸、络氨酸、苏氨酸等等。有趣的是较少有人去尝试使用组氨酸,尽管其侧链具有很好的亲核性,并且该氨基酸存在于很多酶的活性中心。
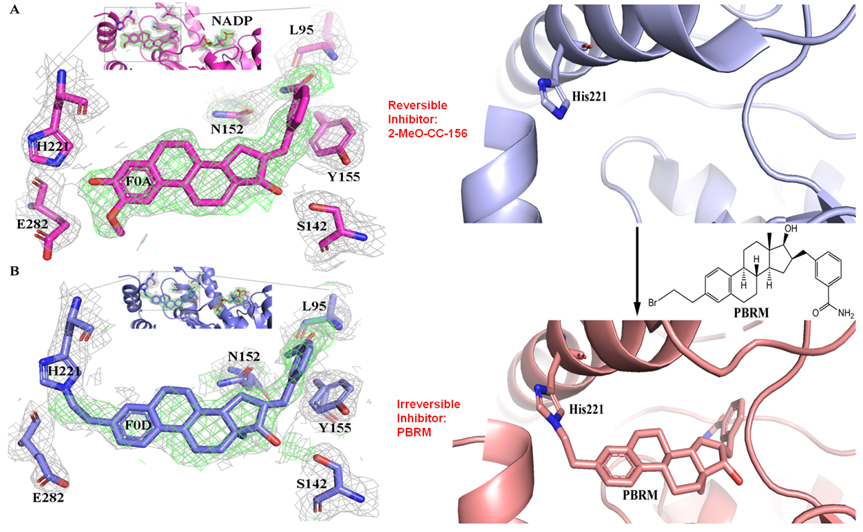
人17β-羟类固醇脱氢酶一型(17β-HSD1)是雌激素活化最为重要的酶之一, 该酶同时具有活化雌激素与降解雄激素的双功能, 是雌激素依赖型疾病治疗的重要靶标。针对该酶的抑制剂的开发已有半个世纪的历史,但由于开发的抑制剂往往具有雌激素性和缺乏足够的选择性,至今还未有该酶的抑制剂进入临床试验。抑制剂PBRM是加拿大拉瓦尔大学CHUL研究中心Donald Poirier教授主导开发的新一代17β-HSD1抑制剂,具有非常高的抑制活性、极高的选择性以及没有内在雌激素性等特性,有望进入临床试验。细胞及分子生物学分析表明该抑制剂具有非可逆抑制剂的特性,但是具体作用机制尚不明确。
李唐博士利用共结晶的方法成功获得了17β-HSD1与PBRM的复合物晶体,结构解析后发现17β-HSD1的His221与PBRM之间形成了共价键,即组氨酸侧链上的氮原子取代了PBRM上的溴原子与碳原子相连。这是目前为止首次报导的组氨酸残基与低反应活性的卤代烷之间发生亲和取代形成共价键的例子。结构分析表明PBRM与酶的底物结合位点附近氨基酸残基之间通过氢键、π键、疏水相互作用等紧密结合,使得PBRM上溴代烷侧链与His221侧链维持在足够近的范围之内,从而促使了该亲核取代反应的发生。该抑制剂的设计为基于低反应活性的卤代烷的高选择性共价抑制剂的开发指明了一条道路。

